En los estudios de bioequivalencia (BE), la pregunta clave no es si dos medicamentos son iguales, sino si puedes confiar en que lo sean. Un error en el cálculo del tamaño de la muestra puede hacer que un medicamento genérico sea rechazado por la FDA o la EMA, aunque sea químicamente idéntico. Esto no es teoría: en 2021, el 22% de las cartas de respuesta completa de la FDA citaron problemas con el tamaño de la muestra o el cálculo de la potencia. Y no es por falta de esfuerzo. Es por falta de precisión.
¿Por qué la potencia y el tamaño de muestra importan tanto en bioequivalencia?
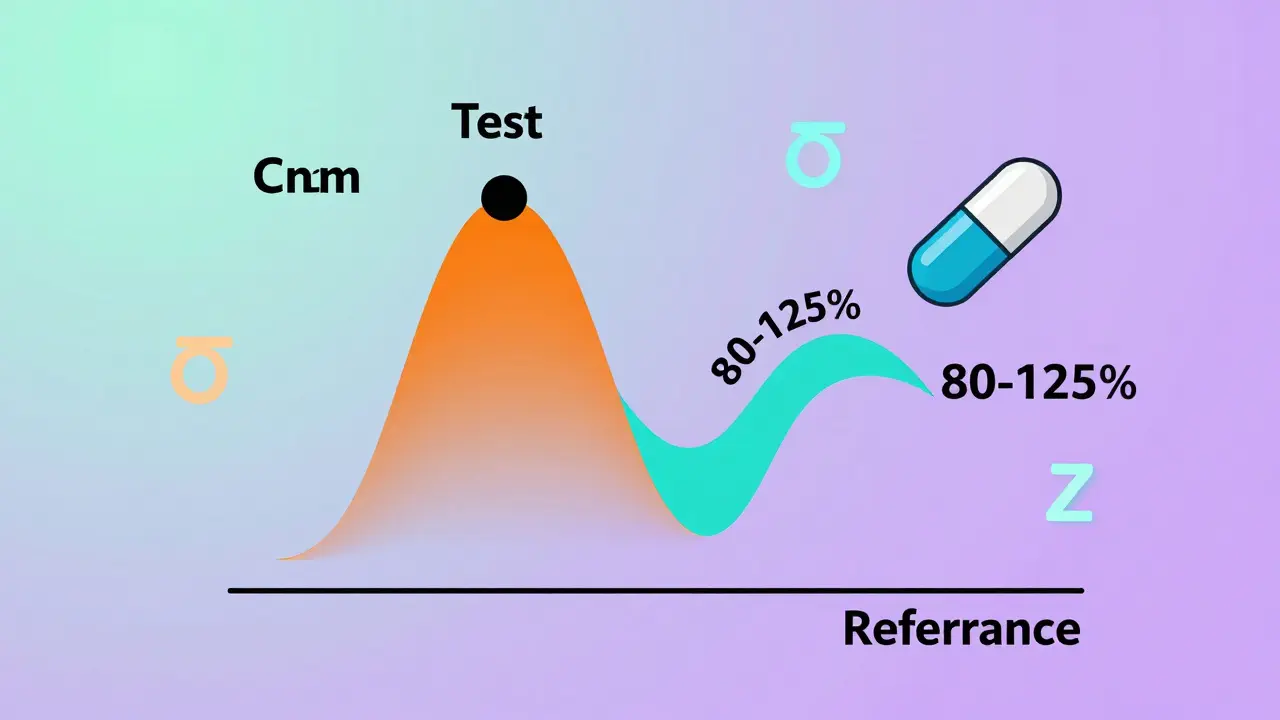
En un estudio de superioridad, quieres demostrar que un fármaco es mejor. En un estudio de bioequivalencia, quieres demostrar que no hay diferencia clínicamente significativa. Eso suena más fácil, pero es más difícil estadísticamente. No puedes simplemente comparar promedios. Tienes que probar que la relación de medias geométricas (GMR) entre el producto de prueba y el de referencia cae dentro de un rango muy estrecho: 80% a 125%. Si la GMR es 1.26, el medicamento no es bioequivalente. Si es 0.92, lo es.
Pero aquí está el problema: los datos farmacocinéticos -como Cmax y AUC- no siguen una distribución normal. Sigue una distribución log-normal. Por eso, todo se analiza en escala logarítmica. Y eso cambia todo el cálculo. Una pequeña variabilidad en la absorción del fármaco puede hacer que necesites el doble de voluntarios. Por eso, la potencia estadística no es un detalle técnico. Es el pilar del estudio.
¿Qué es la potencia estadística y por qué se fija en 80% o 90%?
La potencia (1 - β) es la probabilidad de detectar una diferencia real si existe. En bioequivalencia, no quieres decir que dos productos son equivalentes cuando no lo son (error tipo I, α). Pero tampoco quieres decir que no son equivalentes cuando sí lo son (error tipo II, β). La FDA y la EMA aceptan α = 0.05. Eso significa que hay un 5% de riesgo de falsa aceptación. Pero la potencia varía.
La EMA permite 80% de potencia para la mayoría de los fármacos. La FDA, en cambio, espera 90% para medicamentos de índice terapéutico estrecho, como la warfarina o la fenitoína. ¿Por qué? Porque si un paciente recibe un genérico que no se absorbe bien, puede tener un efecto tóxico o perder el control de su enfermedad. Un estudio con 80% de potencia tiene un 20% de probabilidad de fallar, incluso si el medicamento es perfectamente equivalente. Con 90%, esa probabilidad baja al 10%. Eso no es solo estadística. Es seguridad.
¿Cómo se calcula el tamaño de muestra real?
La fórmula básica para estudios cruzados es:
N = 2 × (σ² × (Z1-α + Z1-β)²) / (ln(θ₁) - ln(μt/μr))²
¿Qué significa esto en la práctica? No necesitas memorizarla. Pero sí necesitas entender qué variables la controlan:
- CV% (coeficiente de variación intra-sujeto): Mide cuánto varía la absorción del mismo paciente de una dosis a otra. Si es 15%, necesitas menos gente. Si es 35%, necesitas casi el doble.
- GMR esperado: La relación que esperas entre el genérico y el original. Si asumes 1.00 (perfecta igualdad) pero el verdadero GMR es 0.95, tu muestra se vuelve insuficiente. En un estudio de 2021, asumir un GMR de 1.00 cuando era 0.95 aumentó el tamaño de muestra requerido en un 32%.
- Márgenes de equivalencia: 80-125% es estándar. Pero para Cmax en algunos fármacos, la EMA permite 75-133%. Eso puede reducir el tamaño de muestra hasta un 20%.
- Diseño del estudio: Un diseño cruzado (mismo paciente, dos periodos) es más eficiente que uno paralelo. Puedes necesitar la mitad de voluntarios.
Imagina esto: un fármaco con CV% de 20% y GMR de 0.95 necesita 26 sujetos para 80% de potencia. Si el CV% sube a 30%, necesitas 52. Si sube a 40%, necesitas más de 100. Eso no es un error de cálculo. Es una realidad farmacocinética.

¿Qué pasa con los medicamentos altamente variables?
Algunos fármacos -como la ciclosporina o el valproato- tienen CV% por encima del 30%. Para ellos, usar el rango 80-125% sería injusto. Por eso existe el RSABE (bioequivalencia promedio escalada por referencia).
El RSABE ajusta los límites de equivalencia según la variabilidad real del fármaco. Si el CV% es 40%, los límites pueden ampliarse hasta 70-143%. Esto reduce el tamaño de muestra de 120 a 30-40 sujetos. La FDA lo permite desde 2018. La EMA lo acepta desde 2010. Pero no es automático. Tienes que demostrar que el fármaco es realmente altamente variable con datos de estudios previos. No puedes inventar un CV% alto para ahorrar dinero.
¿Qué herramientas usan los profesionales?
No se hace con calculadoras de celular. Se usan programas especializados:
- PASS: El más completo. Tiene módulos específicos para BE, RSABE y diseños adaptativos.
- nQuery: Popular en farmacéuticas grandes. Fácil de usar, pero caro.
- FARTSSIE: Gratuito y confiable. Ideal para pequeñas empresas y académicos.
- ClinCalc: Online, gratuito y actualizado. Perfecto para una estimación rápida.
Un estudio de 2022 encontró que el 78% de los estadísticos en la industria usan estas herramientas iterativamente. Cambian el CV%, el GMR, la potencia… y ven cómo cambia el número de sujetos. No lo hacen una vez. Lo hacen cinco veces. Porque la incertidumbre es parte del proceso.

Errores comunes que hacen fallar los estudios
Los estudios no fallan por mala ciencia. Fallan por descuido.
- Usar CV% de la literatura sin validar: La FDA encontró que los valores de literatura subestiman la variabilidad real en un 5-8%. Si usas un CV% de 20% porque lo leíste en un artículo, pero el fármaco real tiene 28%, tu estudio está condenado.
- No considerar la pérdida de participantes: Si calculas 30 sujetos y esperas un 10% de abandono, debes reclutar 33. Si no lo haces, tu potencia cae de 80% a 70%. Y eso puede hacer que rechacen el estudio.
- Ignorar los dos parámetros (Cmax y AUC): Solo calcular potencia para el más variable es un error. La ASA recomienda calcular la potencia conjunta. Solo el 45% de los patrocinadores lo hacen. Pero la FDA lo exige.
- No documentar el cálculo: La FDA pide: software usado, versión, valores exactos, justificación. Si no lo incluyes, el estudio se rechaza por incompleto. En 2021, el 18% de las deficiencias estadísticas fueron por esto.
¿Qué se viene en el futuro?
La bioequivalencia está cambiando. La FDA está promoviendo enfoques modelados, donde se usan simulaciones por computadora y modelos farmacocinéticos para predecir la respuesta. Esto podría reducir el tamaño de muestra hasta en un 50% para fármacos complejos como los de liberación modificada o los biológicos.
Pero hoy, solo el 5% de los estudios usan estos métodos. Porque son nuevos, complejos y no hay guías claras. Por ahora, la regla sigue siendo: calcula bien, documenta todo, y nunca subestimes la variabilidad.
Un estudio bien diseñado no es caro. Un estudio mal diseñado cuesta millones. Y puede retrasar la llegada de un medicamento genérico a los pacientes que lo necesitan. Por eso, cada sujeto cuenta. Y cada decimal en el CV% también.
¿Qué hacer si estás diseñando un estudio de bioequivalencia?
- Reúne datos reales de CV% de estudios piloto. No confíes en la literatura.
- Usa un GMR conservador: 0.95, no 1.00.
- Calcula la potencia para Cmax y AUC juntos.
- Añade un 10-15% extra para pérdidas.
- Usa PASS, FARTSSIE o ClinCalc. Nunca hagas cálculos manuales.
- Documenta todo: software, versiones, valores, suposiciones.
- Si el CV% > 30%, evalúa RSABE desde el inicio.
¿Cuál es el tamaño de muestra mínimo aceptable en un estudio de bioequivalencia?
El tamaño mínimo depende de la variabilidad. Para fármacos con CV% bajo (menos del 10%) y diseño cruzado, 12-18 sujetos pueden ser suficientes. Pero para la mayoría de los fármacos, el rango típico es de 24 a 36 sujetos. Si el CV% supera el 30%, puede llegar a 48 o más. No existe un número fijo. Lo que sí es obligatorio es que la potencia sea al menos del 80% y que el cálculo esté justificado.
¿Por qué se usa la escala logarítmica en los estudios de bioequivalencia?
Las concentraciones de fármacos en sangre (Cmax y AUC) siguen una distribución log-normal, no normal. Esto significa que los datos están sesgados: hay más valores bajos y pocos muy altos. Al transformarlos a logaritmos, se vuelven normales y se pueden analizar con métodos estadísticos estándar como el ANOVA. Además, la relación de medias geométricas en escala original es equivalente a la diferencia de medias en escala logarítmica, lo que permite usar el rango 80-125% de forma válida.
¿La EMA y la FDA tienen los mismos requisitos de potencia?
No siempre. La EMA acepta 80% de potencia para la mayoría de los fármacos. La FDA espera 90% para medicamentos de índice terapéutico estrecho, como anticoagulantes o antiepilépticos. Esto crea desafíos para los genéricos que se quieren comercializar en ambos mercados. Si planeas una presentación global, es más seguro diseñar el estudio con 90% de potencia desde el principio.
¿Puedo usar un estudio previo para justificar el CV%?
Sí, pero con cuidado. La FDA revisó 147 estudios y encontró que el 63% de los CV% tomados de la literatura subestimaban la variabilidad real en al menos un 5%. Si usas datos de otro fármaco, o de otro laboratorio, o de una formulación diferente, puedes estar cometiendo un error grave. Lo ideal es usar datos de un estudio piloto con tu propia formulación. Si no tienes uno, usa el CV% más alto que encuentres en literatura confiable y añade un margen de seguridad.
¿Qué pasa si mi estudio falla por falta de potencia?
Si el estudio no logra demostrar bioequivalencia por baja potencia, no puedes simplemente añadir más sujetos y volver a intentarlo. Eso invalida el diseño. Tienes que reiniciar el estudio desde cero, con un tamaño de muestra calculado correctamente. Esto puede costar más de 500.000 euros y retrasar la aprobación por más de un año. La mejor estrategia es invertir bien en el diseño inicial.



Nahuel Gaitán
enero 10, 2026 AT 10:50Me encanta cómo explicaste lo de la escala logarítmica. Siempre me confundía por qué no se usan promedios normales, pero ahora veo que es porque la farmacocinética es inherently skewed. Es como tratar de usar una regla recta para medir una curva de la vida.
Valentina Juliana
enero 11, 2026 AT 00:21El punto sobre el CV% de la literatura es crítico. En un proyecto reciente, asumimos 18% basado en un paper de 2019, y el estudio piloto mostró 29%. Terminamos reclutando 48 sujetos en vez de 28. La FDA nos pidió la justificación, y al menos no rechazaron el estudio -pero sí nos retrasaron 6 meses. Nunca más sin datos propios.
valentina Montaño Grisales
enero 11, 2026 AT 14:50¡Oye, oye, oye! ¿Y qué pasa con los fármacos de liberación modificada? ¿No deberíamos estar hablando de PK/PD modeling desde el inicio? ¡Esto es 2025! ¿Aún usamos PASS como si fuéramos en los 90? ¡Es como usar un fax para enviar un currículum!
Ana Barić
enero 12, 2026 AT 08:20Me encanta que alguien finalmente hable de esto sin rodeos. Soy farmacéutica clínica y cada vez que veo un protocolo con 24 sujetos y CV% de 15% me entra un sudor frío. ¡No hay fármacos reales con esa variabilidad! Por favor, usen ClinCalc, es gratis y te salva la vida. ¡Y sí, documenten todo, por el amor de Dios!
Isabel Garcia
enero 12, 2026 AT 08:59La EMA y la FDA no son iguales, y eso no es un detalle, es una trampa. Si diseñas para 80% de potencia pensando solo en Europa, y luego quieres entrar a EE.UU., te van a rechazar sin piedad. No hay excusa. Si quieres global, diseña para 90% desde el principio. No es caro. Es inteligente. Y si no lo haces, no es un error estadístico: es negligencia profesional.
George Valentin
enero 12, 2026 AT 09:52Esto que dices sobre el RSABE es interesante, pero no es una solución, es una excusa. ¿Por qué no se fomenta la innovación en formulaciones con menor variabilidad en vez de ajustar los límites? Es como decir que si un coche consume mucho combustible, en vez de mejorar el motor, aumentamos el tamaño del tanque. No resolvemos el problema, lo disfrazamos. La ciencia debería aspirar a la precisión, no a la conveniencia estadística. La industria está poniendo el listón más bajo porque es más barato. Y eso no es ético.
Andrea Fonseca Zermeno
enero 12, 2026 AT 16:34Lo de añadir un 10-15% extra por pérdidas me salvó la vida en un estudio el año pasado. Pensé que 30 era suficiente, no conté con que dos personas se fueron por un accidente familiar. Terminamos con 28 completos, potencia de 76%. Fue un susto. Ahora siempre lo pongo en el protocolo. Gracias por recordarlo.
Gonzalo Andrews
enero 12, 2026 AT 16:57Lo más triste no es que la gente subestime la variabilidad… es que lo haga porque no quiere invertir en estudios piloto. Pero luego, cuando pierden medio millón de euros y un año de trabajo, se quejan. La ciencia no es un juego de azar. Es un compromiso con la precisión. Y si no estás dispuesto a hacerlo bien desde el principio, no deberías estar en este campo. No es agresividad, es verdad.
Sergi Capdevila
enero 14, 2026 AT 16:41¿Alguien ha pensado que quizás la bioequivalencia como concepto está obsoleta? ¿Qué pasa si lo que realmente importa es la respuesta clínica, no el Cmax o el AUC? Tal vez deberíamos dejar de medir fármacos y empezar a medir pacientes. ¿No es eso lo que realmente importa?